L’ anemia mediterranea è una malattia nota anche come anemia eritoblastica o come beta talassemia: come si può intuire, interessa in modo particolare le persone che vivono nell’area del Mediterraneo, ma in realtà si riscontra anche nel Sud Est asiatico, in Iran, nella Cina meridionale e nella Penisola Araba.
L’anemia mediterranea è una malattia con cui si può certamente convivere ma che non va sottovalutata in quanto può avere effetti devestanti e mortali sul paziente se non trattata adeguatamente. Vediamo perché.
Anemia mediterranea : che cosa è
L’anemia mediterranea rappresenta la più comune forma di anemia, il cui effetto negativo principale è rappresentato da una minore produzione di globuli rossi e di emoglobina nel sangue.
Semplificando al massimo possiamo dire che i pazienti affetti da anemia mediterranea producono globuli rossi più piccoli del normale, in numero inferiore rispetto alla normalità. Allo stesso tempo la quantità di emoglobina, che è contenuta all’interno di questi globuli rossi (pochi e piccoli), è inferiore rispetto alla normalità. Per capire bene il problema basta una immagine:
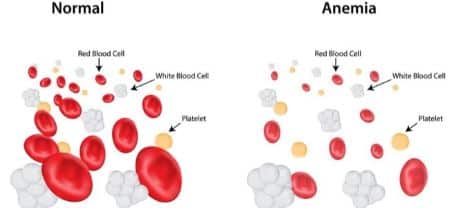
Le conseguenze dovute a un numero ridotto di globuli rossi e a poca emoglobina, possono essere anche gravi, come dicevamo.
Il compito dell’ emoglobina, lo ricordiamo, è infatti quello di trasportare ossigeno all’interno dell’organismo. Quando il nostro corpo non riceve più l’apporto di ossigeno adeguato, iniziamo ad avvertire una spiacevole sensazione di stanchezza. In un uomo in salute, i livelli di emoglobina non dovrebbero scendere sotto i 12 o 13 g/dl a seconda del sesso.
Entrando nel dettaglio, in caso di anemia mediterranea è l’HBa il tipo di emoglobina che risente del disturbo: l’HBa si compone di quattro catene polipeptidiche, due Alfa e due Beta. Le catene Beta sono assenti, o comunque vengono prodotte in misura molto contenuta, nelle persone che soffrono di anemia mediterranea.
Non bisogna, comunque, eccessivamente preoccuparsi: questa patologia che in passato poteva risultare anche mortale, oggi può essere cronicizzata anche nella sua forma più grave attraverso farmaci, trasfusioni e uno stile di vita adeguato.
Anemia mediterranea : le cause
Le cause dell’anemia mediterranea sono solo genetiche: non ci sono virus a cui attribuire la colpa, e di conseguenza non si rischia di essere contagiati nel caso in cui si entri a contatto con persone che ne soffrono.
Tutto dipende dalla mutazione di un gene, l’HBB, che è predisposto alla sintesi della beta-globina, una proteina che fa parte delle proteine adulte di emoglobina.
Il difetto genetico impedisce all’organismo di produrre sub unità di beta globine; la loro totale assenza si indica con il nome di beta-zero (B0).
Altre mutazioni al gene HBB consentono invece una ridotta produzione di beta globuline. In questo caso si tratta di una Talassemia Beta-più (B+).
Sebbene possa apparire inconsueto, la gravità dell’anemia non è strettamente legata alla forma riscontrata: episodi di Talassemia Major(vedi sotto) sono stati riscontrati in soggetti B0 così come in B+, mentre la Talassemia si manifesta in forma intermedia anche in soggetti b0.
Si ritiene che la patologia si sia diffusa soprattutto nel Mediterraneo come risposta alla malaria: una sorta di protezione naturale, se è vero che i globuli rossi delle persone con la beta talassemia immunizzano il corpo umano rispetto alla febbre malarica e contrastano la sua contaminazione.
Anemia mediterranea : tipologie
L’anemia mediterranea è classificata in tre forme diverse in base alla gravità dei disturbi che provoca:
- La talassemia minor
- la talassemia intermedia
- la talassemia major (quest’ultima è nota anche come morbo di Cooley)
Le tre tipologie di beta talassemia sopra esposte sono in ordine di gravità. Ognuna di esse si caratterizza per sintomi differenti, che in alcuni casi sono addirittura assenti.
Chi soffre di talassemia minor, per esempio, non ha sintomi: si tratta della forma di anemia mediterranea più comune, che non provoca problemi di alcun genere nella vita quotidiana. Poiché è impossibile individuarla attraverso la sintomatologia, ci si può accorgere di esserne colpiti solo grazie a un test da laboratorio.
Diverso è il discorso per la talassemia intermedia, che si caratterizza per una sintomatologia molto variabile: in alcuni casi può essere violenta e fastidiosa, mentre in altre circostanze può essere blanda o poco evidente.
Infine, ecco la talassemia major, che è in grado – unica tra le tre tipologie – di influenzare in maniera negativa la qualità della vita nelle sue forme più devastanti.
Chi soffre del morbo di Cooley, in effetti, è costretto a impiegare farmaci ferrochelanti e a ricorrere a frequenti trasfusioni. Trattare la talassemia major è fondamentale sin dal primo momento in cui viene diagnosticata: in caso contrario si rischiano effetti mortali già in età pediatrica.
Un ulteriore problema connesso a questa tipologia di anemia mediterranea è rappresentato dal fatto che a volte non è possibile assecondare il bisogno di trasfusioni a causa di un sovraccarico di ferro, da cui deriva un peggioramento del quadro clinico. Il morbo di Cooley colpisce solo chi eredita il gene difettoso dell’anemia mediterranea da tutti e due i genitori.
Anemia mediterranea, sintomi e classificazione delle sue forme
Ma che significa nel concreto anemia major, minore o intermedia?
Diamo uno sguardo ai sintomi più frequenti e immediatamente riconoscibili delle tre tipologie di anemia.
Talassamia Major o morbo di Cooley
Il morbo di Cooley è la forma più invalidante di Beta Talassemia che necessita di cure e trasfusioni costanti. In alcuni casi la richiesta di trasfusioni non può essere soddisfatta per motivi legati al sovraccarico di ferro e questo complica ulteriormente il quadro clinico.
Per sviluppare la talassemia major è necessario ereditare il gene difettoso da entrambi i genitori. I sintomi della Talassemia Major cominciano ad essere riscontrabili nel bambino fra il primo e il secondo anno d’età.
Uno dei primi campanelli d’allarme è il lento sviluppo del piccolo, cui fa contrasto un ingrossamento delle ossa del cranio, in particolar modo degli zigomi. Le ossa del soggetto con anemia mediterranea Major subiscono lievi o gravi deformazioni, causati dall’espansione del midollo osseo nel tentativo di produrre un maggior numero di globuli rossi.
Un sintomo immediatamente riconoscibile è l’ittero, a causa del quale la pelle e gli occhi assumono un colorito giallastro. Normalmente i genitori che osservano questi cambiamenti nel proprio figlio provvedono a chiedere consulto al medico.
La visita metterà in evidenza un ingrossamento degli organi, quali fegato, cuore ma soprattutto milza, sottoposta ad un sovraccarico di lavoro. I test da laboratorio indicheranno emoglobina hbf in aumento. Se non trattata, la talassemia Major può avere effetti mortali già durante la prima decade di vita.
I sintomi evidenti di talassemia Major sono i seguenti:
- colorito della pelle pallido o giallastro;
- deformità nelle ossa;
- Anemia;
- Improvviso aumento di peso
- Scarso appetito
- Addome ingrossato
- Crisi di pianto
- Irritabilità
- Crescita lenta
- Anormale sonnolenza e senso di stanchezza
- Urine più scure del normale
- Splenomegalia (milza ingrossata).
I sintomi non devono necessariamente manifestarsi tutti insieme.
Talassemia intermedia
In questa condizione i sintomi della malattia sono meno evidenti rispetto alla Talassemia Major, ma l’assenza di proteina beta è comunque sufficientemente importante da compromettere la regolare produzione di emoglobina e globuli rossi, e dunque in seconda istanza da determinare moderata anemia e in alcuni casi significativi problemi di salute.
Le condizioni dei pazienti nati con Talassemia intermedia devono essere valutate dallo specialista sulla base del singolo caso. Talvolta i sintomi possono essere assimilabili a quelli di una talassemia Minor e pertanto rimanere latenti per molto tempo, ma non di rado il confine fra talassemia intermedia e morbo di Cooley è molto sottile.
La discriminante che passa fra un soggetto con Talassemia Major e uno con Talassemia intermedia è – in generale – il numero di trasfusioni cui dovrà essere sottoposto. Un paziente con talassemia intermedia avrà bisogno di trasfusioni per migliorare la qualità della propria vita. Chi è affetto da Talassemia Major invece ne ha bisogno per sopravvivere.
I sintomi della talassemia intermedia:
- Crescita lenta
- Ritardo della pubertà
- Difetti all’apparato scheletrico
- Pallore
I sintomi della talassemia major in forma più blanda
Talassemia Minor
La forma più lieve di anemia mediterranea è quasi sempre asintomatica e non ha ripercussioni sulla qualità della vita.
L’assenza di proteina beta non interferisce in maniera significativa con la normale produzione di emoglobina e, solitamente, gli unici effetti riscontrabili sono una lieve anemia che non necessita di trasfusioni.
Il problema principale per il soggetto cui è stata diagnosticata Talassemia Minor è di natura genetica. Egli è infatti un portatore sano e il suo gene difettoso può essere trasmesso al nascituro.
Questo è un fattore fondamentale da tenere in considerazione per eventuali gravidanze. L’unione con un partner che è a sua volta un portatore sano di beta talassemia definisce un 25% di possibilità di dare alla luce un figlio con talassemia Major. Nel 50% dei casi il nascituro sarà un portatore sano della malattia, mentre per il restante 25% non erediterà alcun gene difettoso.
La talassemia minor viene quasi sempre individuata per caso in occasione di esami di routine. Il diametro dei globuli rossi è di dimensioni più piccole e queste cellule hanno una forma irregolare. A causa di queste caratteristiche, l’anemia mediterranea minore viene è a volte scambiata per anemia sideropenica. Nell’anemia minor gli esami di laboratorio evidenziano un aumento dell’emoglobina di tipo hb 12.
Anemia Mediterranea : diagnosi
La diagnosi di anemia mediterranea, oltre che sulla base dei sintomi, è eseguita mediante esame del sangue. Gli individui con anemia mediterranea hanno i globuli rossi di dimensioni più piccole rispetto alla norma. Queste cellule sono anche numericamente meno presenti e al microscopio palesano forma irregolare e pallore.
Nell’anemia mediterranea minor di solito i parametri di emoglobina hba2 sono elevati. Anche i valori di ferro e ferritina sierica possono essere più alti.
Anemia Mediterranea : diagnosi prenatale
La talassemia può inoltre essere diagnosticata anche con test molecolari. Questi, possono essere eseguiti in qualsiasi momento, inclusi i mesi prima della nascita con un test prenatale, al fine di scoprire se il bambino nascerà talassemico o meno.
I test prenatali effettuati in laboratorio sono:
L’amniocentesi: si effettua fra la sedicesima e la diciottesima settimana di gravidanza – o dopo la venticinquesima (amniocentesi tardiva) -, prelevando il liquido amniotico dalla cavità uterina della madre.
Villocentesi: con questa tecnica invece del liquido amniotico si preleva una parte del tessuto dove sono contenuti i villi coriali. La villocentesi deve essere eseguita fra l’undicesima e la tredicesima settimana di gravidanza.
Anemia mediterranea: trattamento e cura
I tre tipi di anemia che abbiamo visto hanno sintomi di pericolosità differenti e pertanto richiedono cure e trattamenti differenti.
Il trattamento di routine per le anemie più pericolose prevede la trasfusione di sangue da un donatore a un talassemico ricevente. La quantità e la durata dei cicli di trasfusione viene stabilita dallo specialista in relazione alla gravità della Talassemia e alla quantità di ferro libero in circolo nell’organismo.
I talassemici minori non devono essere trasfusi e non devono fare i conti con le controindicazioni che una grave talassemia comporta. Di base, tuttavia, tutti i soggetti talassemici farebbero bene a seguire dei ritmi di vita regolari, a partire dal rispetto di una dieta equilibrata che riduca la quantità di ferro di provenienza animale.
Una conseguenza dell’anemia mediterranea è infatti il sovraccarico di questo minerale nell’organismo. Esso viene “abbandonato” in circolo dai globuli rossi morti precocemente e si accumula per le frequenti trasfusioni.
Per ovviare a questo problema, il trattamento per l’anemia mediterranea maggiore prevede in taluni casi integrazioni di acido folico.
Se nonostante queste precauzioni i livelli di ferro dovessero rimanere alti, potrebbe essere necessario far ricorso alla terapia chelante per ridurre i rischi di tossicosi.
Che cosa è la terapia chelante?
Un agente chelante è una sostanza chimica in grado di chelare – cioè rastrellare – i metalli e le tossine in eccesso nell’organismo.
Nel caso dei beta talassemici quindi il ferro. Il ferrochelante più utilizzato è l’acido etilendiamminicotetracetico (EDTA) che negli Stati Uniti è adoperato da oltre 40 anni.
La terapia chelante viene praticata in particolar modo per l’avvelenamento da piombo e in generale per le gravi intossicazioni.
Il farmaco va somministrato per via sottocutanea tramite microinfusore nei tempi indicati dal medico (in genere circa 12 ore al giorno). Attualmente sono in fase di studio farmaci chelanti orali che renderanno certamente la terapia meno pesante per il paziente.
Tuttavia questo trattamento va attuato soltanto quando è strettamente necessario a quando è lo specialista a ritenerlo indispensabile. Gli agenti chelanti non riconoscono le sostanze “utili” e potrebbero chelare anche esse, rendendo obbligatorio un reintegro. Inoltre la loro espulsione avviene mediante l’urina, pertanto i reni sono chiamati a un lavoro di straordinari.
Trattamento beta talassemia minor
I soggetti con talassemia minor non devono essere trasfusi e possono condurre uno stile di vita normale. I principali accorgimenti da osservare riguardano la dieta, che dovrebbe limitare tutti quegli alimenti ricchi di ferro animale, come il fegato, le frattaglie, le cozze.
Una buona abitudine è quella di accompagnare i pasti con una tazza di te’ al fine di ridurre l’assorbimento del minerale.
Trattamento talassemia intermedia
Sarà esclusivamente il medico, dopo aver valutato il caso specifico, a suggerire alla famiglia o al paziente il trattamento per la talassemia intermedia. Le variabili sono davvero molteplici ed è azzardato tentare di stabilire una cura che sia comune per tutti.
Molto spesso, il medico deciderà di sottoporre il soggetto a trasfusioni solamente quando i livelli di emoglobina scenderanno sotto una certa soglia. Altre volte prescriverà un ciclo di trasfusioni per un determinato periodo di tempo, con l’obiettivo di rivalutare la situazione al termine della cura.
La strategia da seguire è strettamente legata alle condizioni di salute del talassemico e al rapporto fra ferro ed emoglobina nell’organismo; per questo motivo i controlli medici dovranno comunque essere rispettati nei tempi indicati dallo specialista.
Di fondamentale importanza nel trattamento delle forme talassemiche intermedie è che i livelli di emoglobina non scendano mai al di sotto di una soglia minima considerata a rischio.
Trattamento anemia mediterranea major
I bambini con anemia mediterranea risultano in salute fino al primo anno di vita, poiché dotati di un particolare tipo di emoglobina caratteristico del feto e dei neonati. L’emoglobina difettosa viene prodotta con l’avanzare dell’età, e insieme ad essa compaiono i primi sintomi e l’esigenza di un trattamento che li contrasti e scongiuri la morte del piccolo paziente.
Sebbene nei soggetti con anemia mediterranea cronica il sovraccarico di ferro libero sia elevato, l’organismo necessita comunque di trasfusioni croniche.
Il bambino a cui è stata diagnosticata la talassemia maggiore dovrà abituarsi ben presto alle continue trasfusioni di sangue.
Il compito dei genitori è arduo, ma con pazienza il piccolo talassemico imparerà a vedere nel dottore e nella puntura non una punizione ma un sistema per stare meglio.
Rischi dell’anemia mediterranea
I talassemici gravi sono mediamente più soggetti a sviluppare patologie nel corso della loro vita rispetto agli individui in salute. I maggiori pericoli che la beta talassemia porta con se’ sono per il cuore.
Altri organi a rischio sono i polmoni, il fegato e la milza. Possono presentarsi anche problemi all’apparato muscolo scheletrico e il diabete.
Trattamento per i problemi di osteoporosi e osteopenia
Avere un apparato muscolo scheletrico in salute è fondamentale per ciascuno di noi. Le ossa sono l’impalcatura del nostro corpo e poiché esse devono sostenerci per tutta la vita, nostro compito è preservarle.
L’anemia Mediterranea, per i motivi che abbiamo già analizzato, è spesso associata a problemi alle ossa quali osteopenia e osteoporosi. Si tratta di patologie che talvolta rimangono nascoste fino alla manifestazione dei loro primi drammatici effetti: la rottura di un osso in un episodio banale. Prima di incorrere in spiacevoli sorprese, i soggetti con Beta Talassemia dovrebbero eseguire un esame di mineralogia ossea computerizzata finalizzato alla verifica degli indici T-score e Z-score, ossia i parametri di riferimento che definiscono una densità ossea nella norma.
Sia che l’osteopenia e l’osteoporosi siano già diagnosticate, sia nel caso che il talassemico voglia prevenirle, è di fondamentale importanza apportare alcuni accorgimenti alla dieta, prediligendoalimenti ricchi di calcio e vitamina D o gli eventuali integratori.
I fumatori hanno inoltre un motivo in più per provare ad uscire dal tunnel. Le sessioni di esercizi fisici, una tantum, pianificate con un buon fisioterapista possono rappresentare un toccasana.
Nei casi di osteopenia e osteoporosi conclamati, alcuni medici scelgono di intervenire con farmaci bifosfonati come l’acido Pamidronico. Recenti ricerche suggeriscono che l’acido Zoledronico sia utilizzabile nei casi di osteoporosi associati alla Talassemia.
Quante possibilità ho di mettere al mondo un figlio talassemico grave se ho l’anemia mediterranea minore?
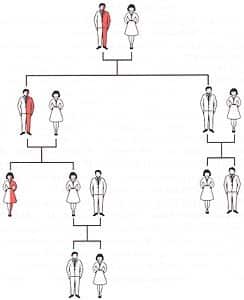
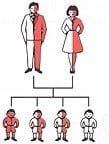
La risposta a questa domanda è strettamente legata alle condizioni del partner. Se uno dei partner è un portatore sano di anemia mediterranea ma l’altro non ha la condizione, non sussiste la possibilità di mettere al mondo bambino con morbo di Cooley. Vi è però il 25% di probabilità che il bambino sia portatore sano a sua volta, come da figura.
Le cose cambiano qualora entrambi i partner abbiano l’anemia mediterranea. In questo caso vi è un 25% di possibilità che il neonato sia talassemico grave, un 25% che non abbia alcun tratto della malattia e il 50% che erediti solamente uno dei tratti difettosi e che dunque nasca con la forma minore di questo male.
Cosa succede se ho l’anemia mediterranea maggiore o intermedia e non faccio la trasfusioni?
L’anemia Mediterranea causa una precoce morte dei globuli rossi. Quando questo avviene, le cellule vengono distrutte nella milza, costringendo l’organo a un sovraccarico di lavoro. La milza dunque si ingrossa e per fronte all’incombente anemia, il midollo osseo incrementerà i suoi ritmi di lavoro producendo maggior emoglobina e un crescente numero globuli rossi, destinati però a fare la fine dei precedenti.
Senza le trasfusioni, Il ferro che i globuli rossi trasportano verrà rilasciato nell’organismo, la milza si ingrosserà (fino a renderne necessaria l’asportazione) e il midollo osseo dovrà sostenere dei ritmi irregolari.
Anemia Mediterranea e trapianto di midollo osseo
L’unica cura “definitiva” contro l’anemia mediterranea è il trapianto di midollo osseo. Esso consiste nella somministrazione di cellule staminali emopoietiche prelevate dal midollo osseo di un donatore compatibile, che si andranno a sostituire a quelle “difettose” del ricevente.
Purtroppo, non sempre questa pratica è attuabile. Presupposto indispensabile affinché si possa procedere al trapianto di midollo osseo è la compatibilità fra donatore e ricevente. Le possibilità di trovare cellule compatibili nel registro mondiale dei donatori sono molto ridotte; fra consanguinei stretti (fratello e sorella), queste aumentano ma sono sempre al di sotto del 30%.
Ci sono diverse controindicazioni che rendono rischioso questo procedimento, dovute alle possibilità di rigetto dell’organismo ricevente e alla reazione del paziente alla prima fase prevista di chemioterapia.
Il medico autorizzerà il trapianto solamente dopo essersi accertato del buono stato di salute indipendentemente dall’anemia. Inoltre, affinché si possa procedere, gli organi non dovranno aver subito già danni.
Anemia mediterranea e fertilità nel morbo di Cooley
L’accumulo di ferro associato all’anemia mediterranea major (morbo di Cooley) può provocare un ritardo nello sviluppo degli organi femminili e maschili, problema che si ripercuote negativamente sullafertilità e quindi sulla possibilità di avere dei figli.
Nei casi più gravi alcuni dei pazienti non raggiungono la pubertà e sviluppano una condizione nota comeipogonadismo.
Nei maschi, la mancanza di testosterone può causare un ritardo nello sviluppo della prostata e dei testicoli. Nella donna, il disturbo si manifesta con un ritardo nello sviluppo delle caratteristiche sessuali secondarie.
Ritardo della pubertà, ipogonadismo e amenorrea sono caratteristiche che fanno capo a un deficit dell’ormone.Tuttavia, minore è la quantità di ferro “libero” nell’organismo, minori sono i rischi che esso si insinui nell’ipotalamo o nell’ipofisi, due strutture situate nel cranio che – senza scendere troppo nel dettaglio sconfinando in campo prettamente endocrino – regolano l’attività ormonale dell’organismo.
Al fine di scongiurare questo pericolo, i medici possono suggerire la terapia chelante prima che il ferro in esubero si accumuli nelle aree a rischio.Negli individui adulti nei quali i problemi di fertilità si sono già manifestati, la terapia ormonale sostitutiva è spesso condotta a termine con successo.
Anemia mediterranea e gravidanza
Ogni coppia, prima di prendere la decisione di mettere al mondo un figlio, pondera attentamente su svariati fattori. Possono essere di ordine sentimentale, economico o personale.
Una donna con anemia mediterranea deve però assicurarsi che il compagno de lei scelto non abbia la stessa patologia genetica, poiché nel 25% dei casi dall’unione di due talassemici nascerà un talassemico grave (morbo di Cooley).
Se per una donna non talassemica la preparazione al parto è una fase molto delicata, per una donna con anemia mediterranea da moderata a grave, essa può addirittura costituire un rischio. Vediamo in cosa consistono questi rischi esattamente.
{kind=link}
– Durante la gravidanza potrebbe essere richiesto un aumento delle trasfusioni, al fine di garantire alla madre e al feto un valore di emoglobina costante e ottimale.
Livelli di emoglobina al di sotto della norma potrebbero avere conseguenze fatali per il feto. Inoltre, durante una gravidanza, il cuore è costretto a un sovraccarico di lavoro del 30% maggiore del consueto.
Per questa ragione, potrebbe essere necessario aumentare il numero di trasfusioni.
– Qualcuno ipotizza che i farmaci chelanti abbiano effetti negativi sulla salute del feto. I medici pertanto tendono a ridurre la terapia chelante.
– Le donne sofferenti di beta talassemia da moderata a grave potrebbero incorrere in problemi cardiaci. Durante la gravidanza, eventuali sintomi sospetti dovranno essere comunicati al cardiologo, che avrà il compito di anticipare l’insorgenza di complicazioni.
– Tutte le donne con osteoporosi – una complicazione frequente nei talassemici gravi – devono assumere un supplemento di calcio e vitamina D.
– Il 10% dei malati di talassemia manifesta ipotiroidismo. Durante una gravidanza il rischio che subentri una condizione di ipotiroidismo aumenta. La terapia ormonale sostituiva garantisce buoni margini di successo nella prevenzione dell’ipotiroidismo.
– La gravidanza e l’accumulo di ferro sono due fattori di rischio nell’insorgenza del diabete.
Questi avvertimenti potrebbero scoraggiare o affliggere una donna con morbo di Cooley desiderosa di avere dei figli. Tuttavia, i moderni progressi medici hanno già dato la possibilità a parecchie coppie talassemiche in tutto il mondo di portare a termine con successo un parto.
Per approfondire:
http://www.humanitas.it/pazienti/malattie-e-cure/sangue-e-midollo-osseo/7975-anemia-mediterranea-o-talassemia+&cd=7&hl=it&ct=clnk&gl=it;
http://www.pieracutino.it/storia-e-distribuzione-geografica-dellanemia-mediterranea/
anemiamediterranea.it
[bbp-single-tag id=332]
Problemi con le analisi cliniche? Invia una domanda nel FORUM: è gratis
Risposta entro 24 ore